Okulær albinisme er en arvelig tilstand, hvor pigmentdannelsen i øjets strukturer er reduceret, mens hud og hår typisk har normal eller næsten normal farve. Tilstanden medfører nedsat syn, nystagmus og øget lysfølsomhed fra fødslen.
Hvad er okulær albinisme?
Okulær albinisme er en genetisk betinget tilstand, der primært påvirker øjnenes pigmentering. I modsætning til okulokutan albinisme, hvor også hud og hår er påvirket, er det ved okulær albinisme hovedsageligt øjnene, der mangler tilstrækkeligt pigment. Pigmentet melanin spiller en afgørende rolle for nethindens normale udvikling og for at begrænse mængden af lys, der når de lysfølsomme celler. Når pigmentet mangler, opstår der en række synsrelaterede udfordringer allerede i spædbarnsalderen.
Sygdomsudvikling
Okulær albinisme skyldes mutationer i gener, der styrer melaninproduktionen i øjets pigmentepitel. Den hyppigste form (type 1, også kaldet Nettleship-Falls type) nedarves X-bundet recessivt, hvilket betyder, at det primært rammer drenge, mens piger typisk er raske bærere. Den mangelfulde pigmentering påvirker udviklingen af makula (foveal hypoplasi), hvilket er hovedårsagen til det nedsatte syn. Desuden ses en abnorm krydsning af synsnervens fibre i chiasma opticum, som påvirker den binokulære synsfunktion.
Forekomst
Okulær albinisme type 1 rammer cirka 1 ud af 50.000 mænd. Tilstanden forekommer på tværs af alle etniske grupper. Kvindelige bærere kan have diskrete tegn som en ujævn pigmentering af nethinden (et såkaldt mud-splattered fundus) uden væsentlig synspåvirkning.
Risikofaktorer
Da okulær albinisme er en arvelig tilstand, er den primære risikofaktor familiehistorie:
- X-bundet arvegang — mødre, der er bærere af mutationen, har 50 % risiko for at få en søn med tilstanden.
- Konsangvinitet — kan øge risikoen for autosomalt recessive former af okulokutan albinisme, der også har øjenmanifestation.
Diagnostik
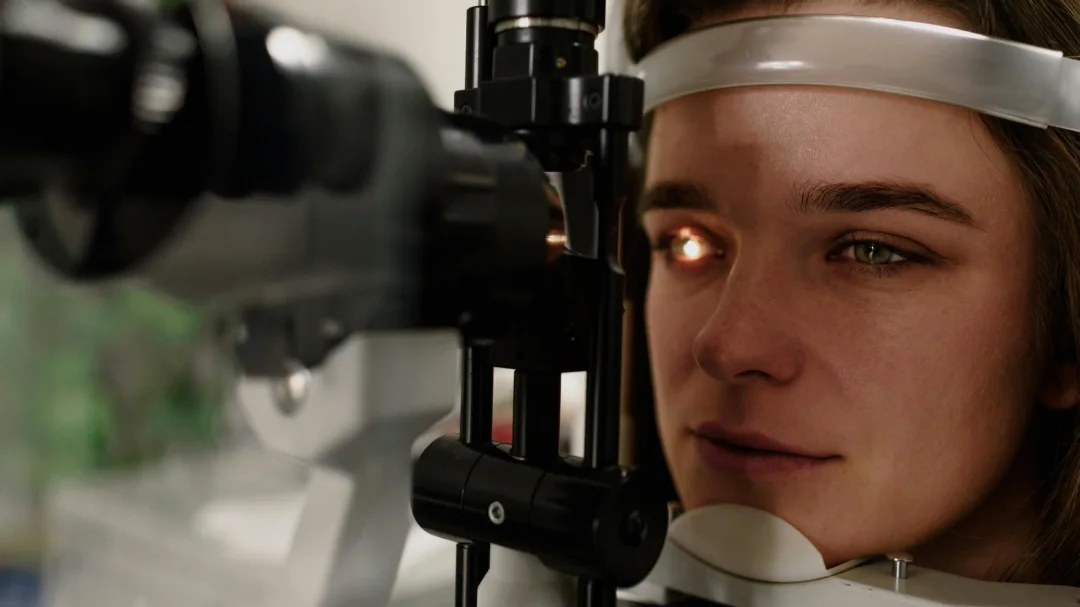
Diagnosen stilles typisk i barndommen på baggrund af det kliniske billede. Ved spaltelampeundersøgelse ses iris-transillumination, hvor lys skinner igennem den tynde, pigmentfattige regnbuehinde. Fundoskopi viser en hypopigmenteret fundus med tydelige choroidale kar og foveal hypoplasi. OCT-scanning kan bekræfte den manglende foveal fordybning. VEP (visuelt evokeret potentiale) kan påvise den abnorme krydsning af synsnervefibre. Genetisk testning kan verificere den specifikke mutation.
Behandling
Der findes ingen helbredende behandling for okulær albinisme. Indsatsen retter sig mod at optimere synsfunktionen og beskytte øjnene:
- Brillekorrektion — mange patienter har betydelig bygningsfejl eller andre refraktionsfejl, der bør korrigeres.
- Solbriller og lysfiltre — reducerer genskin og lysoverfølsomhed.
- Svagsynshjælpemidler — forstørrende hjælpemidler, elektroniske læseapparater og tilpassede skærmindstillinger kan forbedre hverdagens synsfunktion.
- Behandling af nystagmus — i sjældne tilfælde kan prismekorrektion eller kirurgi overvejes for at dæmpe nystagmus.
- Opfølgning hos børneøjenlæge — regelmæssig kontrol sikrer tidlig behandling af eventuel amblyopi og skelen.
Prognose
Synsstyrken ved okulær albinisme er typisk nedsat til mellem 0,1 og 0,3, men varierer fra person til person. Synet er stabilt og forværres ikke med alderen. Med korrekt synsrehabilitering og brug af hjælpemidler kan de fleste patienter leve et normalt og aktivt liv. Det er vigtigt, at børn tidligt får tilpasset undervisningsmiljø og hjælpemidler, så de kan udnytte deres synspotentiale fuldt ud.